Chapter 3 Snakemake Pipeline
The pipeline code is available here. Please refer to this to configure the appropriate working environment.
3.1 Example Data
Here we will take the ATAC-seq / RNA-seq data from this publication as an example to demonstrate our snakemake process and downstream analysis with cisDynet package.
ATAC-seq and RNA-seq data for several immune cell of the human are included here:
| ID | Cells | ATAC-seq | RNA-seq |
|---|---|---|---|
| 1 | Bulk_B | SRR7650729 | SRR7647654 |
| 2 | Mem_B | SRR7650731 | SRR7647656 |
| 3 | Naive_B | SRR7650733 | SRR7647659 |
| 4 | Plasmablasts | SRR7650735 | SRR7647660 |
| 5 | CD8pos_T | SRR7650736 | SRR11007061 |
| 6 | Central_memory_CD8pos_T | SRR7650738 | SRR11007063 |
| 7 | Effector_memory_CD8pos_T | SRR7650740 | SRR11007065 |
| 8 | Naive_CD8_T | SRR7650742 | SRR11007067 |
| 9 | Gamma_delta_T | SRR7650744 | SRR11007069 |
| 10 | Effector_CD4pos_T | SRR7650745 | SRR11007071 |
| 11 | Follicular_T_Helper | SRR7650747 | SRR11007073 |
| 12 | Memory_Teffs | SRR7650749 | SRR11007075 |
| 13 | Memory_Tregs | SRR7650751 | SRR11007077 |
| 14 | Naive_Teffs | SRR7650753 | SRR11007079 |
| 15 | Regulatory_T | SRR7650755 | SRR11007083 |
| 16 | Th1_precursors | SRR7650757 | SRR11007085 |
| 17 | Immature_NK | SRR7650763 | SRR11007193 |
| 18 | Mature_NK | SRR7650764 | SRR11007090 |
| 19 | Monocytes | SRR7650767 | SRR11007092 |
| 20 | pDCs | SRR7650770 | SRR11007095 |
3.2 Config File
All you need to do is prepare a config.yaml file in the following format.
sample:
Bulk_B:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650729_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650729_2.fastq.gz
Mem_B:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650731_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650731_2.fastq.gz
Naive_B:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650733_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650733_2.fastq.gz
Plasmablasts:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650735_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650735_2.fastq.gz
CD8pos_T:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650736_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650736_2.fastq.gz
Central_memory_CD8pos_T:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650738_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650738_2.fastq.gz
Effector_memory_CD8pos_T:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650740_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650740_2.fastq.gz
Naive_CD8_T:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650742_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650742_2.fastq.gz
Gamma_delta_T:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650744_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650744_2.fastq.gz
Effector_CD4pos_T:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650745_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650745_2.fastq.gz
Follicular_T_Helper:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650747_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650747_2.fastq.gz
Memory_Teffs:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650749_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650749_2.fastq.gz
Memory_Tregs:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650751_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650751_2.fastq.gz
Naive_Teffs:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650753_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650753_2.fastq.gz
Regulatory_T:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650755_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650755_2.fastq.gz
Th1_precursors:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650757_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650757_2.fastq.gz
Immature_NK:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650763_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650763_2.fastq.gz
Mature_NK:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650764_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650764_2.fastq.gz
Monocytes:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650767_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650767_2.fastq.gz
pDCs:
fwd: /mnt/public3/cat/data/ATAC/fq/SRR7650770_1.fastq.gz
rev: /mnt/public3/cat/data/ATAC/fq/SRR7650770_2.fastq.gz
## Pipeline working derectory.
workdir: /mnt/public3/cat/data/ATAC/
## BWA indexed genome fasta file.
fasta: /public/workspace/zhutao/pipeline/genome/hg19/bwa_clean_index/hg19_clean.fa
## The gff file.
gff3: /public/workspace/zhutao/pipeline/genome/hg19/gencode.v19.annotation.gff3
## Effective genome size for MACS2 peeak calling.
effect_genome_size: 2864785220
## Motif file with PWM format.
motif: /mnt/public3/cat/data/ATAC/jaspar.moitf.txt
## Sequence_adapters.
adapters: /public/workspace/zhutao/miniconda3/pkgs/trimmomatic-0.39-1/share/trimmomatic-0.39-1/adapters/NexteraPE-PE.fa
## mapping quality tags.
mapping_quality: 30
## Organelle name (For plants, a comma separates mitochondria and chloroplasts)
organell_chr: chrX
## Peak calling.
extsize: 150
shift: 75
## convert bam to bigwig.
smoothLength: 20
binSize: 5
threads: 5
## Remove blacklist from the peaks.
blacklist: NA
overlap_ratio: 0.3 ## Requiring the overlap region being at least xx% of peak as a blacklist.
## The software path.
software:
atacqc: "/mnt/public3/cat/data/ATAC/atacqc"
trimmomatic: "~/miniconda3/bin/trimmomatic"
bwa: "~/miniconda3/bin/bwa"
bedtools: "/bioapps/rhel7/bedtools-2.30.0/bin/bedtools"
samtools: "~/miniconda3/bin/samtools"
macs2: "~/miniconda3/bin/macs2"
bamCoverage: "~/miniconda3/bin/bamCoverage"
fadix: "~/miniconda3/bin/faidx"
multiqc: "~/miniconda3/bin/multiqc"
TOBIAS: "~/miniconda3/envs/tobias/bin/TOBIAS"
3.3 Blacklists
In the context of epigenomic analysis, “blacklisted regions” typically refer to specific areas on the genome that often include repetitive sequences, transposons, genes encoding non-epigenetic modifying enzymes, etc. These regions can potentially interfere with the accuracy and interpretability of epigenomic studies. In mammals, such as humans and mice, the ENCODE programme has obtained specific blacklists by inferring and hand-correcting. This same problem exists in plant epigenomics, but there is no specific blacklist for researchers to use. Here we used the greenscreen software here, combined with data collected by our ChIP-Hub, to infer genome blacklists for Arabidopsis, rice, maize, tomato, and soybean.
The corresponding blacklists can be downloaded via the links below.
| Plant species | Tax ID | Common name | Genome release version | Genome size (in Mb) | # BAMs used |
|---|---|---|---|---|---|
| Arabidopsis thaliana | 3702 | Mouse-ear cress | TAIR10 | 119.15 | 1533 |
| Oryza sativa | 39947 | Japonica rice | IRGSP-1.0 | 374.47 | 140 |
| Zea mays | 4577 | Maize | AGPv3 | 2066.43 | 154 |
| Solanum lycopersicum | 4081 | Tomato | ITAG2.4 | 823.94 | 28 |
| Glycine max | 3847 | Soybean | Wm82.a2.v1 | 949.18 | 53 |
3.4 Pipeline Output Folders
Important Folders:
trimmed: After removing sequencing adapters and low quality sequences.
signal: The bigwig file normalized by CPM and RPKM methods.
q30: The bam file after removing the low mapping scores and organelle reads.
peaks: The narrowpeak files obtained by MACS2.
multiqc: The cisDynet pipeline report.
cut_sites: The Tn5 cuts files after shifting the reads.
ATACCorrect: Bias correction of ATAC-seq reads in open chromatin using TOBIAS
BINDetect: Estimation of differentially bound motifs based on scores, sequence and motifs using TOBIAS.
3.5 Report Explanation
After running snakemake, we can get the appropriate results as well as an html report. Here is a example report.
Duplication rate: The PCR duplication rate.
FRiP: Fraction of all mapped reads that fall into the called peak regions, i.e. usable reads in significantly enriched peaks divided by all usable reads.
Mapping rate: The percentage of reads mapping to genome.
Organell mapping rate: The percentage of reads mapping to Organelle sequence.
Q30 reads number: The number of reads after removing low-quality mapping(Default 30).
Raw reads number: The number of raw reads.
TSS enrichment: Tn5 cuts enrichment scores around the TSS. See ENCODE for details.
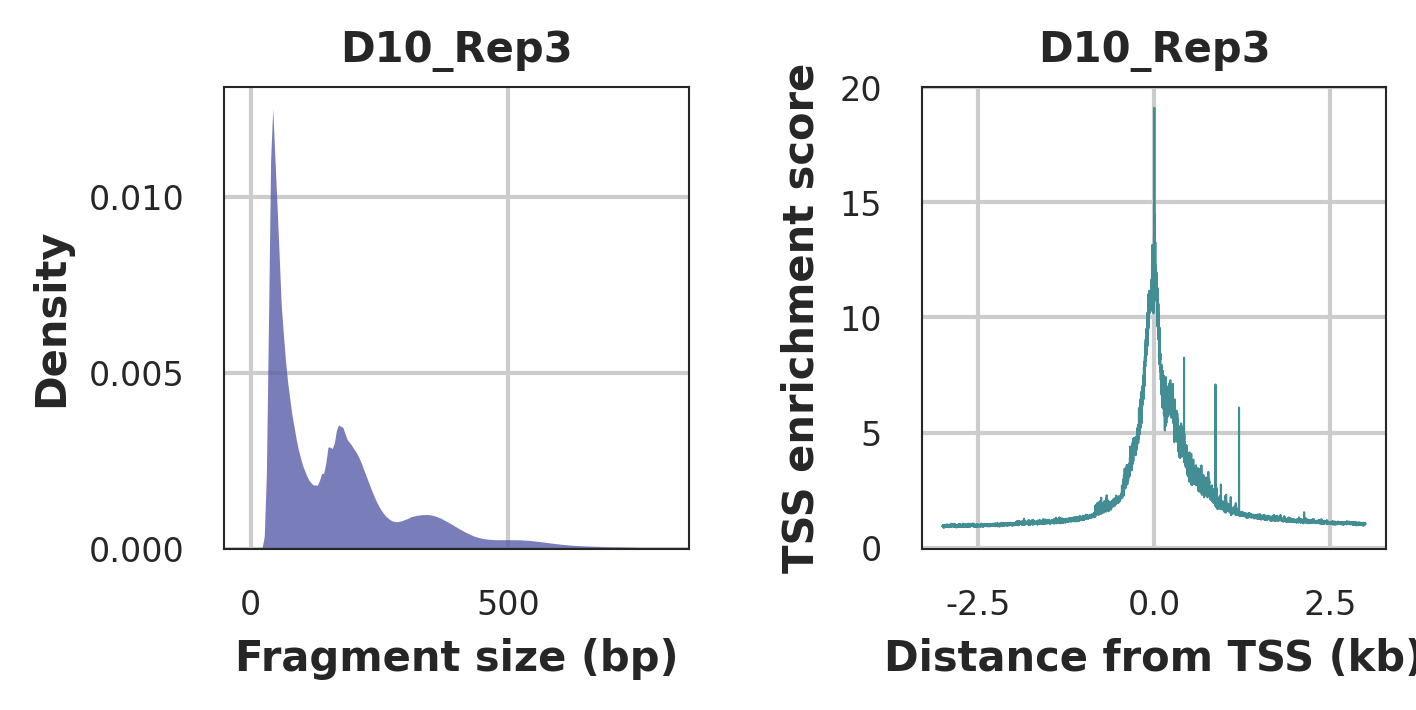 Left: Typical fragment size distribution plot shows enrichment around 100 and 200 bp, indicating nucleosome-free and mono-nucleosome-bound fragments.
Left: Typical fragment size distribution plot shows enrichment around 100 and 200 bp, indicating nucleosome-free and mono-nucleosome-bound fragments.
Right: Typical TSS enrichment plot shows that nucleosome-free fragments are enriched at TSS.
3.6 Troubleshooting
- Low mapping rate (usually < 80%): This may indicate the possibility of microbial contamination during the experiment.
- High organelle mapping rate (usually > 20%): It suggests that a pure suspension of nuclei was not successfully obtained, and additional washes are recommended. It is advised to aspirate as much supernatant (which contains mitochondria) as possible before proceeding with the transposition reaction. In the case of plant cells, the use of flow cytometry for nuclei sorting is highly recommended to minimize organelle contaminants.
- High duplication rate (usually > 30%): This could result from insufficient library complexity, an inadequate number of cells, a low DNA concentration at the start of PCR, or an excessive number of PCR amplification cycles. It is recommended to optimize the Tn5 reaction system or reduce the number of amplification cycles.
- Low TSS enrichment scores (usually < 5): This might indicate issues with the sample, such as the presence of dead cells, floating DNA fragments in the suspension, or severe nucleus damage prior to the Tn5 transposition reaction.
- Low FRiP (usually < 15%): If the experiment is fine, it is essential to verify that the sequencing depth is sufficient.